Examples
eBDIMS simulations have been run for several model proteins and can be accessed from the example page.
Note that:
- each example provides public access to input files and results and can be used as inspiration/template to prepare new input data,
- the list of examples contain the model proteins with rich ensembles that were all thoroughly described and discussed in the original eBDIMS research article (Orellana et al. 2016)
Pathway projection evaulation criteria
By default, the eBDIMS server selects the best two PCs or NMs for projection, i.e. the first two PCs, which typically capture >70% of the structural variance, or the two NMs better overlapped with the modelled transition, which ideally should capture >80% of the movements involved (Orellana et al. 2016, Orellana et al. 2010).
Whether these axes adequately capture the motions of interest to understand a transition, needs to be ultimately judged by the user taking into account biological information about the structures. As a rule of thumb, when several distinct experimental conformations are available, PCs are better descriptors than NMs, which typically only track accurately simple rigid-body harmonic motions.
Note also that NMs computed from different reference structures can be remarkably different (e.g. those computed from open conformations usually track better large-scale transitions). Thus, when analysing transition projections, the user needs to consider two important features:
- Whether the 2D-projection is effectively separating distinct known functional states onto different clusters, e.g. structures bound/unbound, solved in similar conditions, etc. If a pair of motion axes group together structures solved in similar conditions (bound to certain ions or ligands, solved at the same pH, etc) or representing the same state (open/closed, inactive/active, bound/unbound, etc) indicates they are relevant to analyze the transition.
- Whether the 2D-projection of the eBDIMS path is smooth: since each eBDIMS frame necessarily resembles the preceding and the following one, and all of them progress towards a target structure, in a significant motion axis they will project following a smooth line. Backtracking, sharp changes in direction or zigzag paths indicate the axis does not capture adequately the features of the structures.
The features of the projected structures should change gradually along significant modes e.g. rMSD versus the target or start structure, collective variables such as pore radius, key interresidue distances, etc. In general, the default modes presented by the server are the most representative. However, in the case of complex transitions where small-scale higher modes are involved, or whenever the presented axes do not provide smooth projections and neat clustering, exploration of other modes can be useful:
- For PCs, modes beyond the 2nd mode can still capture significant variance, and thus be relevant to further understand/classify the motions driving the transition; in general, a PC with variance > 10% should be explored
- For NMs, lower-energy modes can provide better projections than other modes of higher frequency, which can display marginally better overlap with the transition
Use cases
Below are discussed different model proteins that demonstrate the different eBDIMS applications: basic path sampling, path assymetry, intermediate assignment, and monitoring of MD simulation.
Basic path-sampling (Ribose-binding protein)
Ribose-binding protein (RBP) is a periplasmic protein that displays a large hinge motion of two similar domains upon ligand binding. The 11-mer ensemble covers the entire closing process including intermediates.
Note that PC1 describes most of ensemble variance (~98%) as interdomain rotation, which distinguishes three structure clusters along the forward eBDIMS pathway projections (blue). In this example, NM1 accurately tracks the conformational change (overlap ~0.83) and thus can also identify the intermediates as such, although grouped into two instead of three clusters.
Note also that while an interpolation would project as a single straight line between start and target conformers, the two eBDIMS paths are slightly divergent and enclose an area of the conformational landscape, which we have shown correspond to the sampling by atomistic MD simulations (see details in Orellana et al. 2016).
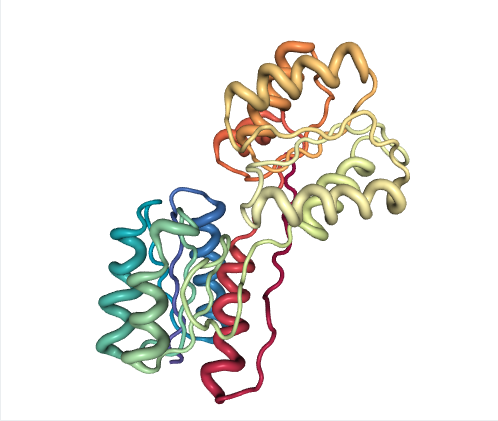
Figure 1: Ribose-binding protein (RBP)
Path assymetry (RNAse)
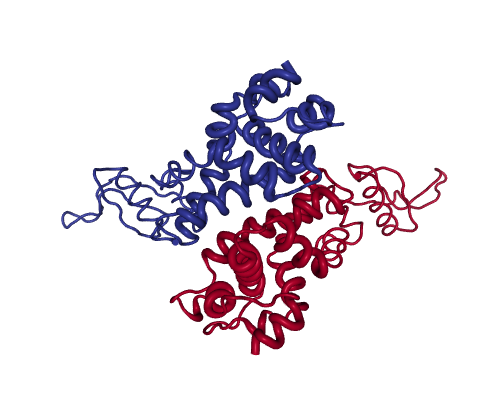
Figure 2: RNAseIII
RNAseIII is a Mg2+-dependent homodimer that modulates gene expression by cleaving dsRNA. The catalytic site is formed by a highly charged cleft between the two subunits.
The PCs of the experimental 11-mer ensemble decompose the motions associated to catalysis into parallel (PC1) and antiparallel (PC2) rotations of the two dsRNA binding domain “arms”, which cover 52% and 43% variance. In absence of Mg2+, dsRNA binds to dsRBDs “arms” outside the cleft (1YYO structure, projected at 0,0 in PC-space). Upon Mg2+ binding, the arms can reorient dsRNA to the cleft (1YYW, projected at -251, 247).
Both NM and PC projections show significant asymmetry for the forwards and reverse pathways, which suggest sequential protein motions. In this example, NMs have low overlap with the transition (0.40, 0.33) and thus cannot partition structures efficiently. However, the X-ray ensemble contains two groups of intermediates approached along the forward and reverse transition, which are evident upon projection onto the main PCs: along the forward pathway, eBDIMS visits one single structure (1YZ9, -219, 125), while the reverse path departs and significantly approaches a cluster of structures (0, 300) solved in presence of Mg2+.
Intermediate assignment (SERCA)
The SERCA pump is an intensely studied P-type ATPase that transports Ca2+ ions across cell membranes, with more than 50 structures solved bound to ligands and ATP analogues, covering nearly all of the catalytic cycle. The PCs tracking the opening and rotation of intracelullar Ca2+-binding domains allow to project this large ensemble in a way which allows immediate visualization of the motions connecting the different catalytic stages: on the right, projections show the transition from open (0,0) to closed (80,-400) Ca2+-bound structures upon ATP binding; then movement to the left cluster (-300,-150) across PC1 corresponds to ATP hydrolysis coupled to ion pumping. Ensemble PCA along with pathway projection shows that inhibitors like sarcolipin (4hw1, near 0, -100) or certain mutations trap intermediate conformers along the forward transition from the Calcium-bound open state to the Calcium-ATP bound closed state. As observed for RBP, the two eBDIMS paths contain most of the area sampled by MD simulations (more details in Orellana et al. 2016).
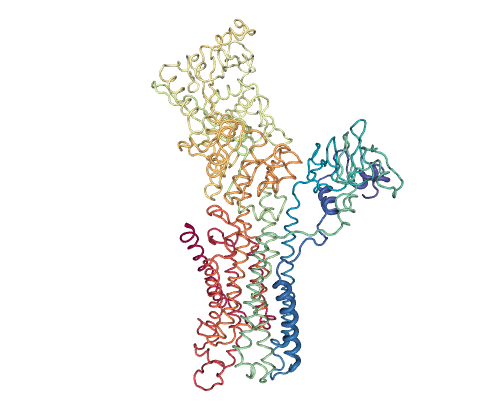
Figure 3: SERCA
Monitoring of MD simulation (GLIC)
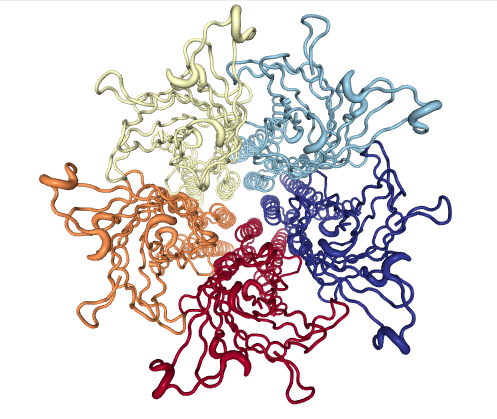
Figure 4: GLIC
GLIC is a prototypic member of the pentameric ligand-gated ion channels family, which plays a central role in neurotransmission through the opening of their ion-conducting pore upon ligand binding.
The 45-mer GLIC ensemble tracks the transition from the closed (0,0) to open channels, passing through a series of locally closed structures. Projections allow visual evaluation of the sampling by a MD simulation of GLIC at channel opening conditions (pH4); note how a simulation starting from the closed state (4npq) approaches the locally closed intermediate area but does not reach the cluster of fully open structures (4hfi and others). As in Example 1, the eBDIMS paths delimit most of the area sampled by MD. More details in Orellana et al.